

Clinical characteristics
In total, 254 children were included in the kidney transplantation registry from 2013 to 2022. Of these, 244 (males: females 1.6:1) enrolled in the study with written consent. All 244 patients received first-time graft transplantation, at a median age of 13.1 years. They were followed for a median of 2.2 years (interquartile range [IQR], 1.5–4.7 years), resulting in 791.6 person-years of follow-up. The majority of this time (657 years, 97.5%) was spent with a functioning transplant. Phenotypic profiling revealed that the initial clinical diagnoses included SRNS/nephritis (108/244, 44.2%), CAKUT (36/244, 14.8%), kidney cystic disease (24/244, 9.8%), Alport syndrome (6/244, 2.5%), tubulopathy (3/244, 1.2%), Fabry disease (1/244,0.4), and kidney failure of unknown etiology (KFu, 66/244, 27.0%) (Fig. 1). Biopsy-based diagnosis was registered in 20 patients. Extrarenal phenotypes were observed in 52 patients, including hearing loss (n = 16), cardiological disorders (n = 6), neurological disorders (n = 5), visual loss (n = 3), achromatopsia (n = 1), nystagmus (n = 1), and short stature (n = 21). In total, 18 probands had a family history of kidney disorders.
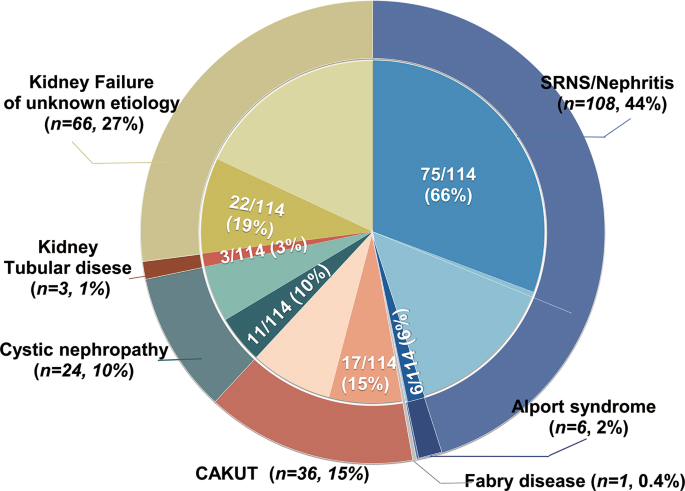
Distribution of clinical diagnosis and post-exome diagnosis in 244 pediatric kidney transplant recipients. The outer circle represents the numbers and percentages of transplant recipients who were classified into one of five clinical diagnostic groups: steroid-resistant nephrotic syndrome (SRNS) or nephritis (process blue), Alport syndrome (satin silver), Fabry disease (navy blue), congenital anomalies of the kidney and urinary tract (CAKUT, dark maroon), kidney cystic disease (cade blue), tubulopathy (saddle brown), and kidney failure of unknown etiology (KFu, pale goldenrod). Inner segments represent for each diagnosis group the relative fraction of patients in whom a final gene diagnosis was confirmed post exome sequencing (dark color) or the unsolved patients in whom pathogenic variants in monogenetic disease-causative genes were identified (light color)
A monogenic cause is identified in 46.7% of kidney transplant recipients
ES was conducted for all families with the exception of 19; parental samples were unavailable for one of these 19 families. The average sequencing depth for all samples was 146X (range 97–215). More than 95% of designed exonic regions were sequenced 20 times for all samples (QC metrics for all samples are provided in Supplemental Table 1). No instances of consanguinity were observed in any of the families. Known variants of pathogenic genes from the registration records were confirmed in 58 cases. None of the variants that had been diagnosed previously were missed with sequencing.
ES provided a molecular genetic diagnosis for the 114 (46.7%) probands with monogenic variants in 15 known disease-causing genes. Pathogenic or likely pathogenic variants of six genes accounted for 36 (16.1%) patients with autosomal dominant diseases, 19 genes accounted for 62 (25.4%) patients with autosomal recessive diseases, and two genes accounted for 16 (6.6%) genetic diagnoses of X-linked recessive diseases (Supplemental Table 2). Of the 115 diagnosed pathogenic or likely pathogenic variants, 52 were missense, 34 were frameshift or nonsense, eight were CNVs, 19 were canonical splice-site variations, and two were indels (Fig. 2). In 7% of the probands (17/244), we detected variants of uncertain significance in a gene known to cause kidney disease (Supplemental Table 3). The five genes COL4A5, COQ8B, NPHP1, PAX2, and WT1 accounted for 66.9% of the monogenetic kidney disease diagnoses (Fig. 3).

Landscape of the frequency of genes and mutation patterns identified in the pediatric kidney transplant cohort. An oncoplot shows all of the disease-causative genes across our cohort of 114 children with kidney failure. Mutation types and frequencies are summarized for each gene on the right and the mutational burden for each case is shown at the top
Providing a precise etiologic diagnosis for kidney transplant recipients
The percentages of patients for whom we established a molecular genetic diagnosis varied across the clinical diagnostic groups (Fig. 1). ES confirmed the suspected clinical diagnosis in 74/244 (30.3%) of cases and revised the pre-exome clinical diagnoses in 40/244 (16.4%) of cases, including establishing a specific underlying cause for kidney failure in 19 patients with KFu (Fig. 2; Table 1).

Sankey diagram of the trajectories between initial clinical diagnoses, genetic diagnosis, disease-causative genes, and clinical implementation. Left and middle: division of the initial clinical diagnosis and post-exome diagnosis and monogenic disease-causative genes. Middle and right: genetic diagnosis and clinical implementation. The width of the lines in the Sankey plot is proportional to the relative quantity of cases within each group
Among the patients with glomerulopathy, including SRNS and nephritis, monogenic podocytopathies were identified in 49 patients (COQ8B [n = 18], WT1 [n = 9], PAX2 [n = 6], TRPC6 [n = 5], NPHS1 [[n = 2], NPHS2 [n = 2], PLCE1 [n = 1], NUP107 [n = 1], NUP85 [n = 1], NUP93 [n = 1], LMX1B [n = 1], LAMB2 [n = 1], INF2 [n = 1]). In patients with an a priori clinical diagnosis of glomerulopathy, pathogenic variants in COL4A5 or COL4A3 were detected in eight individuals, confirming the diagnosis of Alport syndrome. A family history of nephrosis was reported in all seven families with an Alport syndrome diagnosis. Alport syndrome was identified in an additional seven families who were initially diagnosed with either nephrotic syndrome or nephritis. Three of these families included multiple affected individuals. Genetic findings that modified the diagnosis in 19 patients included mutations in patients with NPHP (NPHP1 [n = 4], TTC21B [n = 3], ANKS6 [n = 1], NPHP3 [n = 1], NPHP4 [n = 1]), collagenopathies (COL4A5 [n = 7], COL4A3 [n = 1]), and Fabry disease (GLA [n = 1]).
Among the patients with CAKUT, pathogenic variants were detected in four known disease-causing genes, including PAX2 (n = 5), EYA1 (n = 2), and SALL1 (n = 1). One patient clinically diagnosed with CAKUT had pathogenic variants in the gene NPHP1. Pathogenic variants were detected in ciliopathy genes NPHP1 (n = 5) and NPHP3 (n = 2) in the seven patients with cystic kidney disease. For another two patients with an initial diagnosis of tubulopathy, the genetic diagnosis was confirmed with NPHP1 and NPHP4, respectively.
Among the patients who developed kidney failure without a known etiology, we confirmed the monogenetic kidney disorders in 11 known disease-causative genes, including PAX2 (n = 4), COQ8B (n = 4), NPHP1 (n = 3), WT1 (n = 2), COL4A5 (n = 1), COL4A4 (n = 1), ANKS6 (n = 1), TTC21B (n = 1), SLC34A1 (n = 1), and SALL1 (n = 1).
Clinical implementation of genetic diagnosis
It takes us about three weeks to perform the WES and to provide the results to the transplant team. Multidisciplinary team would give a consult for each case before transplant surgery. Genetic testing has three main applications in clinical kidney transplantation: risk assessment of donors and family counseling, identification of combined therapy schemes for recipients with genetic etiology, and improvement of post-transplant surveillance (Table 1; Fig. 2). Supplementary Table 2 provides detailed information.
First, the final molecular diagnosis allowed for genetic counseling of the patients’ family members and a full assessment of the living donor candidates. After obtaining informed consent, genetic screening for living donors was conducted in 35 families (Table 1). Reproductive counseling was also provided for 18 families with confirmed genetic diagnoses.
Second, genetic tests provided crucial information for targeted therapies in 24 recipients, which could affect graft function or survival following transplantation. For example, it was necessary to continue the pharmacological treatment of enzyme replacement therapy (ERT) following transplantation for patients with Fabry disease. This provided the clinical clue to close follow-up of vasculitis problems, such as cardiopathy. For the 22 patients diagnosed with Coenzyme Q10 (CoQ10) deficiency-associated glomerulopathy caused by the pathogenic variants of COQ8B, oral supplementation with CoQ10 should be continued following transplantation. And one case with the pathogenic variant of SLC34A1 continued the treatment for osteoporosis.
Third, genetic analysis improved post-transplantation surveillance for 41 children (Supplementary Table 2). In cases with a final diagnosis of syndromic kidney disease (PAX2, EVA1, SALL1, NPHP1, NPHP3, NPHP4, TTC21B, ANKS6, COL4A5, COL4A4), more details were added to the surveillance program, including ophthalmological, otorhinolaryngological, and psychomotor development evaluations during childhood and adolescence. Among the seven patients with identified COL4A5 variants who were initially diagnosed of FSGS, three children developed into KF without hearing impairment or ophthalmological abnormalities. These three patients with variants in COL4A5 (p.Gly435Ar; p.Pro856GlnfsTer19; p.Gly51Arg) need further surveillance for hearing or vision problems as well. For the 11 cases diagnosed with WT1-related nephropathy, the decision to perform prophylactic nephrectomy was based on the genetic identification of WT1 mutations supporting the potential risk of malignancy. The median age of prophylactic nephrectomy was 9.3 years old (IQR, 5.7–13.5 years old). Cancer surveillance was routinely conducted in these patients following transplantation. No complications after nephrectomy was reported.
We emphasize the role of surveillance in cases even without a definitive molecular diagnosis, such as the recurrence of FSGS after kidney transplantation. In the 40 patients (34.8%) referred for FSGS or SRNS, the genetic diagnosis failed to establish this, which could indicate a high risk of post-transplantation recurrence. Nonetheless, among the 75 patients with a definitive molecular diagnosis for SRNS or glomerulopathy, there were no reported cases of proteinuria recurrence during the median follow-up of 2.0 years after kidney transplantation.

Add Comment