
GH128 and CBM6 fold as a whole
The crystal structure of CBM6E was determined at a resolution of 2.4 Å. The structure of CBM6E comprises two distinct domains: an (α/β)-barrel catalytic domain (residues 84–348) belonging to the GH128 family, and a C-terminal β-sandwich domain (residues 349–473) belonging to the CBM6 family (Fig. 1a). The CBM6 domain forms an interdomain interface with GH128 primarily through hydrogen bonds between α8 and α9 of the GH128 domain and β10 and β13 of the CBM6 domain (Fig. 1b). Notably, the interdomain interface involves the following residues: D86, K351, R88, Y350, N289, H369, R296, A361, S338, E359, F348, and H362, with each pair forming hydrogen bonds. Additionally, R88, F348, Y350, M288, V339, and A379 participate in hydrophobic interactions as trios (Fig. 1c). Using the ConSurf web server, we identified that most of the residues at the interdomain interface are evolutionarily conserved (Additional file 1: Fig. S1). This conservation suggests a certain degree of evolutionary stability in the folding of GH128 and CBM6 as a cohesive unit.

Overall structure of CBM6E. a Domain architecture of CBM6E. The CBM6E protein contains a tandem arrangement of two domains: GH128 and CBM6. SP, signal peptide. b Overall structure of the tandem GH128 and CBM6 domain of CBM6E. The catalytic GH128 module adopts an (α/β) barrel scaffold, where the α-helices are depicted in magenta and the β-sheet in cyan. On the other hand, the CBM6 module adopts a common β-sandwich fold, which is colored yellow. c Interdomain interface between GH128 (cyan) and CBM6 (yellow). Residues that participate in forming contacts between the two domains are represented using a stick representation. Hydrogen bonds, indicated by dashed lines, are formed between specific residues with a distance of less than 3.5 Å
Structural comparison
We conducted a comprehensive structural comparison of CBM6E with entries in the Protein Data Bank (PDB) using the DALI server. The analysis revealed that the closest structural neighbors belonged to subgroup II of the GH128 family: ScGH128_II (PDB code 6uax) and PvGH128_II (PDB code 6uav) [20]. ScGH128_II and PvGH128_II exhibited sequence identities of 42% and 39%, respectively, with the GH128 domain of CBM6E. Regarding structural similarity, ScGH128_II displayed an RMSD value of 1.5 Å over 241 matched Ca atoms, while PvGH128_II had an RMSD value of 1.7 Å over 247 matched Ca atoms. In addition, this analysis indicated that the CBM6 domain of CBM6E shared similarities with ZgLamCCBM6 (PDB code 5fui), a marine CBM6 module known for its ability to bind laminarin [21]. The structural alignment revealed an RMSD value of 1.5 Å over 117 matched Ca atoms, accompanied by a sequence identity of 22%. Notably, we observed a Mg2+ ion within CBM6E-CBM6, occupying a similar position as seen in ZgLamCCBM6 (Additional file 1: Fig. S2a). This ion demonstrated an six-coordinate octahedral geometry and coordinated with Glu357 OE1, Glu359 OE1, Ala379 O, Asn467 OD, Asn467 O, and a water molecule (Additional file 1: Fig. S2b). The Mg2+ complex was supported by its six-coordinate octahedral geometry and B factors consistent with neighboring atoms. Furthermore, upon modeling a Ca2+ ion, which similarly adopted a six-coordinate octahedral geometry, into the same site, it led to a negative (red) Fo–Fc map for the ion after crystal structure refinement (Additional file 1: Fig. S2c).
This DALI search also yielded some tandem GH and CBM modules, such as Geobacillus stearothermophilus β-d-xylosidase XynB1 (PDB code 2bfg) [22] and Pseudomonas aeruginosa PslG (PDB code 4zn2) [23]. However, the relative orientations between the GH and CBM domains in these proteins totally differ from what was observed in CBM6E. The distinct directional arrangement of the GH and CBM domains may not only influence the stability of the protein comprising these domains but also play a crucial role in the coordinated recognition of specific polysaccharide substrates by these domains, a phenomenon suggested for certain multimodular glycoside hydrolases [24]. Additionally, there are instances where the binding of a CBM to a carbohydrate ligand and the accommodation of a substrate in the active site of the catalytic module do not exhibit coordinated movements, often characterized by flexible interdomain orientations [24, 25].
Negative-subsite region
The crystal structure of CBM6E complexed with GOS was obtained through cocrystallization of the E168Q inactive mutant of CBM6E with laminarihexaose (L6) and determined at a high resolution of 1.28 Å. This complex structure enabled the mapping of two β-1,3-GOS chains within the catalytic groove of the GH128 domain: one spanning from the -4 to -1 subsites and the other extending from the + 1 to + 5 subsites (Fig. 2a). The cleavage of the glycosidic bond between the − 1 and + 1 subsites might be attributed to residual enzyme activity of the E168Q mutant enzyme at high crystallization concentrations. The elongated catalytic groove of CBM6E aptly accommodates the curved conformations of the two GOS chains, which collectively form a single-helical structure (Figs. 2b and 7c). This structural characteristic correlates with CBM6E’s preference for linear curdlan over branched laminarin, as well as its distinctive cleavage pattern yielding products L3 to L6.

Substrate recognition within the GH128 catalytic domain of CBM6E. a Illustration of the interactions between active-site residues and β-1,3-glucooligosaccharides (GOS). GOS molecules with degrees of polymerization (DPs) of 4 and 5 are depicted in magenta and orange, respectively, representing their positions within the negative- and positive-subsite regions. The insets show expansions of the negative- and positive-subsite regions, as well as the region neighboring the + 5 subsite, respectively. Residues engaged in interactions with GOS are depicted as cyan-colored sticks, while residues neighboring the + 5 subsite, selected for tyrosine substitution, are portrayed as blue-colored sticks. b A polder map contoured at 2σ is presented for GOS with DPs of 4 and 5, observed within the negative and positive-subsite regions, respectively, as derived from the crystal structure of the E168Q inactive mutant of CBM6E-GH128/CBM6 cocrystallized with laminarihexaose. Additionally, CBM6E is represented using an electrostatic potential surface depiction
The interactions within the negative-subsite region are primarily governed by two conserved hydrophobic residues: W141, spanning from − 4 to − 2, and W117 at the − 2 subsite. At the − 4 subsite, D142 forms hydrogen bonds with the glucosyl C4-OH and C6-OH, occupying the position of water molecules observed at the − 5 and − 4 subsites in AmGH128_I, a member of the GH128 subgroups I (Additional file 1: Fig. S3a and b). The glucoside at the − 2 subsite forms hydrogen bonds with Q174. The glucoside at the − 1 subsite establishes hydrogen bonds with N167, Q168, H245, and E272. Additionally, F310 stacks against the glucosyl moiety at the − 1 subsite. Furthermore, a β-configuration is trapped within the − 1 subsite (Fig. 2b), which aligns with the retaining mechanism observed in the GH128 family. The conserved residue Y247 serves as a nucleophile-activating element, while E168 and E272 function as catalytic acid residues.
Positive-subsite region
In the reported complex structures of subgroup II of the GH128 family, no glucosyl moieties were observed in the positive-subsite regions. We were fortunate to observe glucosyl moieties from + 1 to + 5 in the complex structure of CBM6E (Fig. 2a and c). The + 1 glucoside interacts with aromatic residues F171, Y247, and W276, which correspond to L117, Y192, and W218 of PvGH128_II, respectively (Additional file 1: Fig. S3d). Additionally, Q168 forms hydrogen bonds with the + 1 glucosyl C2-OH and C3-OH. N206 (corresponding to N152 of PvGH128_II) forms hydrogen bonds with the + 1 and + 2 glucosyl C2 hydroxyl groups, as well as the glycosidic bond between the + 1 and + 2 glucosides. D277 forms a hydrogen bond with the + 2 and + 3 glucosyl C2 hydroxyl groups. The + 3 glucoside stacks against Y207 and M248. Y207 corresponds to W153 in PvGH128_II and Y137 for the + 2 subsite in AmGH128_I (Additional file 1: Fig. S3c). Furthermore, W256 and Y250 (corresponding to S198 and Y194 of PvGH128_II, respectively) provide aromatic platforms for the + 4 and + 5 glucosides, respectively. These observations provide additional confirmation that CBM6E is classified within subgroup II of the GH128 family, given its stronger resemblance to PvGH128_II in the positive-subsite region compared to AmGH128_I.
Rational design at the substrate-binding groove
To enhance the production of high-DP GOS, we pursued the extending of the substrate-binding groove by incorporating aromatic residues close to the + 5 subsite based on the CBM6E:GOS complex structure (Fig. 2a). While S252Y displayed approximately a twofold reduction in activity, with a specific activity of 1.9 U/mg compared to the wild-type (WT) protein’s (4.3 U/mg), both E251Y and D293Y exhibited approximately a threefold increase in activity toward curdlan high-set gel compared to the WT, with specific activities of 13.6 U/mg and 11.6 U/mg, respectively (Table 1).
Thin-layer chromatography (TLC) revealed that the E251Y variant generated a more diversified DP of GOS ranging from 3 to 9 (Fig. 3a). Conversely, the D293Y variant yielded relatively pure L5 from curdlan, as depicted by the results (Fig. 3b).
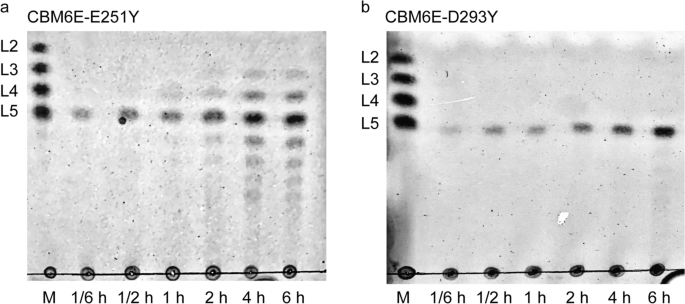
TLC patterns of hydrolytic products of curdlan high-set gel catalyzed by the E251Y (a) and D293Y (b) variants of CBM6E
Molecular docking study using a triple-helical β-1,3-glucan
The enhanced specificity of CBM6E toward curdlan high-set gel compared to curdlan powder suggests that CBM6E has a preference for the triple-helical conformation of curdlan. To identify ancillary binding sites for curdlan triple helices, we conducted molecular docking experiments using a triple-helical β-1,3-glucan undecamer and the apo structure of CBM6E. Two binding models (Model 1 and Model 2) were generated and depicted in Fig. 4 and Additional file 1: Fig. S4, both exhibiting a ZDock Score of 11.7.

Two best-ranked docking poses of triple-helical curdlan to CBM6E. Surface complementarity (a, c) and hydrogen bonding interactions between triple-helical β-1,3-glucan and CBM6E (b, d) are indicated for both simulated structures
In Model 1, the triple-helical β-1,3-glucan displayed a favorable structural complementarity with the CBM6 domain, while its nonreducing ends made contact with the GH128 domain (Fig. 4a and Additional file 1: Fig. S4a). Notably, this model showed binding of the triple helix substrate to a site spanning both catalytic and CBM domains. In this model, the interaction between the triple-helical β-1,3-glucan and CBM6E was facilitated by the formation of six hydrogen bonds. These hydrogen bonds involved the main chain atoms of Q85, Q108, and T440, as well as the side chain atoms of Q108 and T376 (Fig. 4b). Additionally, hydrophobic interactions occurred between the glucan and CBM6E, mediated by residues S131, P353, M439, and S442 (Additional file 1: Fig. S4b).
Model 2 revealed the triple-helical β-1,3-glucan spanning across the active groove of the GH128 domain (Fig. 4c and Additional file 1: Fig. S4c). In this model, a total of fourteen hydrogen bonds were observed between the triple-helical β-1,3-glucan and CBM6E. These hydrogen bonds involved the main chain atoms of Y98, S314, and L328, as well as the side chain atoms of Y96, E173, Q174, N211, S315, E316, and D325 (Fig. 4d). Furthermore, hydrophobic interactions were identified between the glucan and CBM6E, with residues Y98, R313, and S315 playing a role in these interactions (Additional file 1: Fig. S4d).
Overall, both docking models, in conjunction with the crystal structure of CBM6E in complex with GOS, support the hypothesis that triple-helical curdlan unwinding precedes its hydrolysis within the catalytic groove.
Mutational analysis of potential ancillary binding sites for triple-helical curdlan
To evaluate the functional roles of these potential ancillary binding sites in accommodating triple-helical curdlan, we introduced alanine substitutions for all surface-exposed residues (Y98, Q108, E173, S315, E316 in the GH128 domain, and T376 in the CBM6 domain) among them (Additional file 1: Fig. S5a) and subsequently conducted enzymatic kinetics assays. The results are summarized in Table 2 and illustrated in Additional file 1: Fig. S6.
The data from the WT, Y98A, Q108A, and E173A variants were fitted well using the Hill model, as opposed to the Michaelis–Menten model. Notably, all Hill coefficients for both the WT and these mutant CBM6E enzymes exceeded 1, suggesting the existence of two or more binding sites and positive cooperativity in substrate binding. The Y98A, Q108A, and E173A variants displayed K0.5 values of 7.7, 8.5, and 7.9, respectively, which are comparable to the WT’s K0.5 value of 7.6 mg/mL, indicating minimal impact on substrate binding by these mutations.
In contrast, the data from the S315A, E316A, T376A, S315A/T376A, and E316A/T376A mutants did not converge when using the Hill model; hence, a simple Michaelis–Menten model was employed instead. For comparison, the WT data were re-fitted using the Michaelis–Menten model. The S315A and E316A variants displayed slight increases in Km values, measured at 20.4 and 18.2 mg/mL, respectively, compared to the WT’s Km value of 16.7 mg/mL. Conversely, the T376A variant exhibited an even higher increase in Km value, reaching 24.1 mg/mL, indicating a reduced apparent affinity for the substrate due to this mutation. Furthermore, the Km values for the combined variants S315A/T376A and E316A/T376A could not be determined due to saturation levels being too low for accurate extrapolation. Nevertheless, it is anticipated that these values are higher than the highest substrate concentrations (> 25 mg/mL), indicating substantially diminished affinities of these combined variants for curdlan high-set gels.
Collectively, these findings suggest that T376 within the CBM6 domain serves as a crucial ancillary binding site for triple-helical curdlan, while the GH128 and CBM6 domains work synergistically in recognizing curdlan.
T376 in CBM6 represents a novel sugar-binding residue
Interestingly, the T376 residue in CBM6E-CBM6 is located outside the conventional sugar-binding clefts typically observed in CBM6 family members, as demonstrated by the structural alignment of CBM6E-CBM6 with ZgLamCCBM6 (Additional file 1: Fig. S5b). On the other hand, I382 and V435 in CBM6E-CBM6 correspond to Y291 and W348 in ZgLamCCBM6, respectively, which form the aromatic clamp in the variable loop site (VLS), previously referred to as cleft A. V388 in CBM6E-CBM6 corresponds to W297 in ZgLamCCBM6, a critical sugar-binding residue found in the concave face site (CFS), previously known as cleft B.
To assess the contribution of I382, V435, V388, and T376 to curdlan binding, we conducted pull-down assays and determined the dissociation constant (Kd). The V435A mutant displayed a similar affinity for curdlan high-set gels as the WT, with a Kd of approximately 0.9 mg/mL. The T376A, V388A, and I382A variants of CBM6E-CBM6 exhibited approximately 27-, 21-, and 3-fold diminished binding affinity to curdlan high-set gels, with Kd values of 24.4, 18.8, and 3.0 mg/mL, respectively (Table 3 and Additional file 1: Fig. S7). A fluorescent thermal shift assay demonstrated that the T376A mutation had minimal impact on the observed melting temperature (Tm) of CBM6E-GH128/CBM6, ruling out the possibility that the T376A mutation affects the overall folding and stability of CBM6E (Additional file 1: Fig. S8). These findings suggested that T376 in the CBM6 module represents a novel sugar-binding residue, which likely emerged during the coevolution of this CBM module with its appended GH module.
CBM6 binding disturbed curdlan gel’s network structure
Certain CBMs have demonstrated the ability to disrupt the surface of tightly packed polysaccharides, such as cellulose fibers [26, 27] and starch granules [28, 29], leading to the loosening of the substrate and increased exposure to the catalytic module for enhanced degradation. To investigate whether the binding of CBM6E-CBM6 could induce disruption of curdlan gel’s ordered structures, both Congo red staining and SEM assays were performed.
In comparison to curdlan incubated with the negative control BSA, curdlan incubated with the CBM6E-CBM6 WT protein exhibited a notable hypsochromic shift from 497 to 492.5 nm, with a shifting distance of approximately 4.5 nm. This shift indicated a reduction in the ordered structures of curdlan. Conversely, curdlan treated with the binding-deficient mutant T376A of CBM6E-CBM6 exhibited no shift in comparison to curdlan incubated with BSA (Additional file 1: Fig. S9a). Crucially, this observed wavelength shift cannot be attributed to potential interaction disparities between Congo red and distinct proteins. This is substantiated by the absence of any shift in the absorption spectra when comparing Congo red incubated with both the WT and T376A mutant proteins, conducted without the addition of curdlan (Additional file 1: Fig. S9b).
Moreover, the SEM images of the control sample containing pure curdlan gel revealed a consistent and tightly knit network structure with porous features (Fig. 5a and b). In contrast, after treatment with the CBM6E-CBM6 WT protein, noticeable disruptions emerged within the previously uniform curdlan gel structure, resulting in the formation of larger cavities (Fig. 5c and d). Conversely, the introduction of the binding-deficient mutant, CBM6E-CBM6-T376A, did not have a conspicuous impact on the network structure of the curdlan gel (Fig. 5e and f). These observations suggest that CBM6E-CBM6 effectively enhances the enzymatic hydrolysis of curdlan, not only by targeting but also by disruptive effects.

SEM visualization of microstructures in curdlan gel with various treatments. a, b Pure curdlan gel; c, d curdlan gel treated with CBM6E-CBM6-WT protein; e, f curdlan gel treated with the binding-deficient mutant protein, CBM6E-CBM6-T376A. The magnifications of each group images are set as 5000× and 10,000×
Effect of CBM6 addition on enzymatic hydrolytic activity against curdlan
As the CBM6 module has the potential to disturb the compact structure of the curdlan gel, it may facilitate greater access for β-1,3-glucanases to the densely packed β-1,3-glucan chains. Consequently, this could lead to an enhancement in the activities of curdlan-degrading β-1,3-glucanases. In our investigation into the impact of CBM6 addition on curdlan hydrolysis, we specifically focused on two distinct β-1,3-glucanases: a GH128 enzyme, CBM6E, and a GH64 enzyme, KfGH64.
When CBM6E-CBM6 was introduced in various ratios with curdlan gel (ranging from 1:200 to 1:20, and the respective ratios with the CBM6E enzyme varied from 1:4 to 2.5:1, w/w), it led to a remarkable two- to threefold increase in CBM6E activity compared to the negative control, where separate CBMs were absent. Particularly, at a CBM6:curdlan gel ratio of 1:1 (the corresponding ratio with the CBM6E enzyme was 50:1, w/w), the CBM6 incubation resulted in an approximately 4.5-fold increase in CBM6E activity (Fig. 6a).
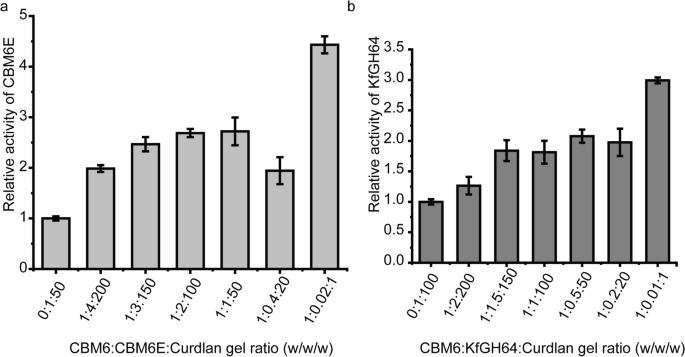
Increasing the CBM6 concentration during curdlan gel hydrolysis results in an increase in enzymatic activity. CBM6E-CBM6 was introduced in various ratios with curdlan gel (ranging from 1:200 to 1:1, w/w) before the enzyme was added into the mixture. The results are expressed as relative activities of CBM6E (a) and KfGH64 (b) against curdlan high-set gel in the absence and presence of CBM6E-CBM6. The consistent enzyme-to-curdlan gel mass ratios were maintained at 1:50 for CBM6E and gel, and 1:100 for KfGH64 and gel
Similarly, upon the addition of the CBM6 to the KfGH64-curdlan reaction mixture with different CBM6-to-curdlan gel ratios (ranging from 1:150 to 1:20 and the respective ratios with the KfGH64 enzyme varied from 1:1.5 to 5:1, w/w), we observed an approximate twofold increase in the rate of reducing sugar release. Notably, at a CBM6:curdlan gel ratio of 1:1 (the corresponding ratio with the KfGH64 enzyme was 100:1, w/w), an approximately threefold increase in the rate of reducing sugar release was achieved (Fig. 6b). These findings suggest that the promotive effect of the CBM6 on curdlan hydrolysis appears to be a consistent phenomenon across various β-1,3-glucanases.
Add Comment